分子模拟
分享关于MD的一切
分子模拟是现代化学和物理学的重要组成部分,尤其是对于大分子的研究。它们已经发展成为成熟的方法,可以有效地用于理解各种大分子体系结构与性质的关系。
3月22日,清华大学化工系燕立唐教授课题组发表研究论文。无论是分子模拟领域入门者还是从事分子模拟相关方向的研究人员,这篇论文都将是一份极好的教程资料。
其旨在厘清分子模拟发展历史以及主要模拟技术,模拟软件如何选择,最后结合四大案例阐述分子模拟技术在大分子科学体系的应用。
通讯作者
燕立唐,2007年在清华大学化学工程系获得工学博士学位,2011年5月回国加盟清华大学化工系,现任长聘副教授。
主要从事大分子及其粒子界面行为的理论计算与模拟研究工作:
通过发展和运用多尺度计算模拟方法和理论模型,紧密围绕复杂拓扑大分子及其各向异性粒子在软物质体系界面上的作用、组装与动力学开展基础理论研究。
☆原文PDF后台回复“教程”获取。
PS:本篇推送涉及的干货知识较多,篇幅较长,可选择感兴趣的部分按需阅读。
分子模拟 历史概述
计算机模拟的发展有着悠久而丰富的历史。最初,电子计算机被利用于第二次世界大战期间的核武器和密码破解的发展。
1952年3月,Metropolis公司对在洛斯阿拉莫斯MANIAC上尝试尽可能广泛的问题感兴趣,这可能是计算机模拟应用于各个学科的起源。
随着计算机技术的发展,通过分子模拟来表征化学物质的静态和动态性质已经成为可能,这革新了研究化学问题的手段。
图1:第一台电子计算机Los Alamos MANIAC和CPU及GPU示意图
由于技术的限制,算法和程序只能由研究人员编写,在分子模拟的早期阶段应用CPU进行计算。
到目前为止,已经开发了各种软件包来同时使用系统中的CPU和GPU,与仅基于CPU的程序相比,提供了极高的性能。
观察和理解都是科学所必需的。在分子模拟问世之前,主要依靠近似的理论是理解和预测分子物质性质的唯一途径。
图2:实验、模拟和理论研究是三种势在必行的方法
分子模拟的好处是,可以用来验证现有理论,同时也为新理论的发展指明了方向。特别是当实验由于条件苛刻而昂贵或具有挑战性时,模拟是实验室研究的重要补充。
因此,实验、分子模拟和理论研究是观察和理解物理和化学问题的三个重要手段。
模拟技术 软件选择
研究者简要介绍了被广泛用于研究和预测大分子的结构、性质的各种模拟和计算技术。可以分为三个主要类别:量子化学计算、原子模拟和粗粒化模拟。
1、量子化学计算及软件
在量子化学计算中,主要考虑电子的运动和影响。第一性原理计算是量子化学计算的代表性方法之一,它将电子自由度和核自由度分开考虑。
该方法只需要原子的核电荷和少数模拟的环境参数作为输入,所有的性质都是通过求解薛定谔方程得到的,不需要使用任何经验参数。因此,使用第一性原理计算的优点是结果更准确,干预更少。
然而,由于计算效率低,人们提出了一种新的方法,即密度泛函理论(DFT),它是基于第一原理计算的。
GAUSSIAN和VASP都是用于量子化学计算的成熟软件包。有一款名为Material Studio的商业Windows/Linux软件也包含一个模块。
该模块可以让用户通过量子化学计算研究气相、溶剂、表面和固体环境中的问题。不精通Linux或编程的研究人员可以使用这个软件。
2、原子模拟及软件
经典的原子水平的分子模拟已经成为提高我们对材料科学和生命系统中分子过程理解的最流行的方法之一。这类方法可以解决许多与大分子的纳米和纳秒尺度相关的任务。
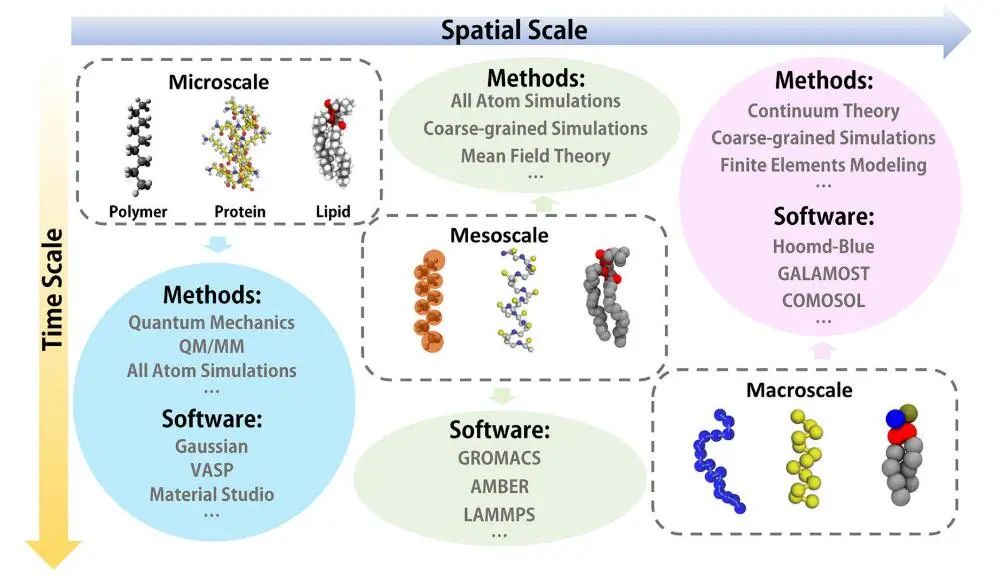
图3:多尺度方法示意图
全原子模拟是一种非常强大的以原子分辨率呈现信息的方法,因为在原子模拟中,一个粒子代表一个特定的原子。但由于目前的计算能力,它只能用于模拟微尺度系统。
针对各种体系和性质创建的最流行的全原子模拟FF有:AMBER FF主要用于研究蛋白质、核酸、多糖等生物分子。
CHARMM力场可以处理各种大分子,包括有机分子、聚合物、生化分子等。这种FF得到的结构、构型能和自由能往往与实验值吻合。
OPLS-AA FF被开发用于有机液体、稀水溶液、氢键和离子水复合物的计算,并提供了一套描述晶体或水溶液中蛋白质非键相互作用的函数。
原子尺度分子动力学模拟的软件包主要包括Material Studio、GROMACS、LAMMPS和NAMD等。MD仿真可以通过修改控制脚本、定义参数、使用命令启动模拟运行来实现。
3、粗粒化模拟
由于计算量随粒子数呈指数增长,使用大量原子来体现所有分子细节非常耗时。在大分子模拟中,一种典型的用于访问长时间尺度的模拟是粗粒化模拟。
在CG模型中,一个粒子代表至少包含几个原子或化学基团的流体区域,这个区域在分子尺度上很大,但在宏观上仍然很小。
与原子模拟相比,CG模拟具有简单、高效的特点,是研究介观尺度性质和揭示普适分子机制的绝佳工具。
四大应用 典型案例
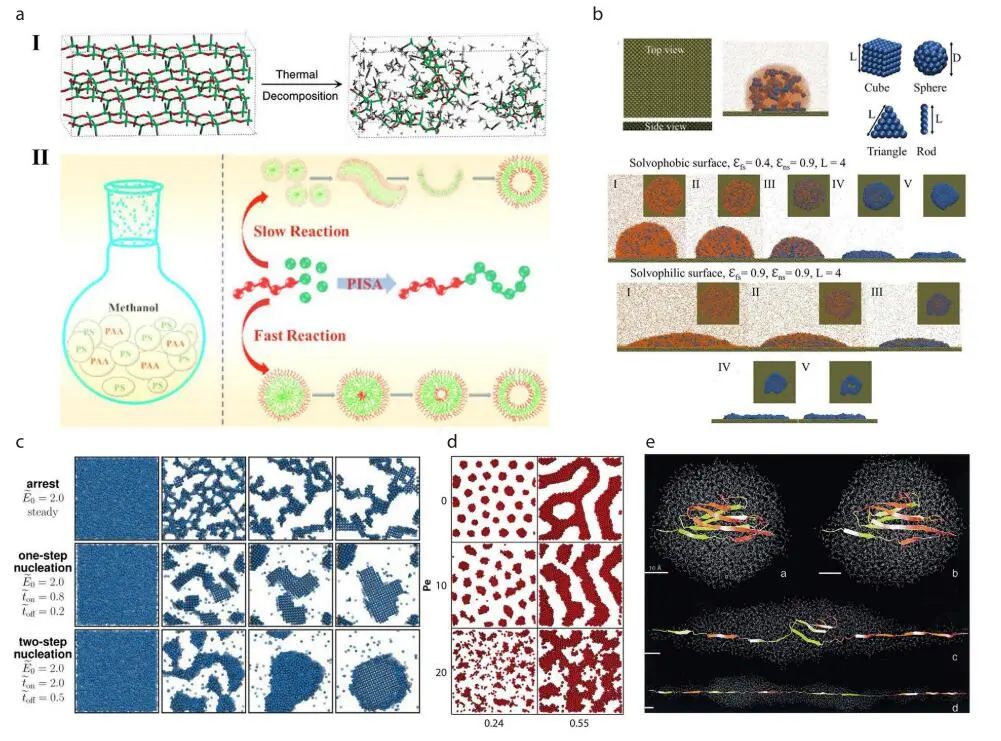
1、自组装
自组装在许多系统中自然发生,是创建纳米结构集合体的重要实用策略。它是在没有人为干预的情况下,各组成部分自主地组织起来,从无序走向有序模式或结构的过程。
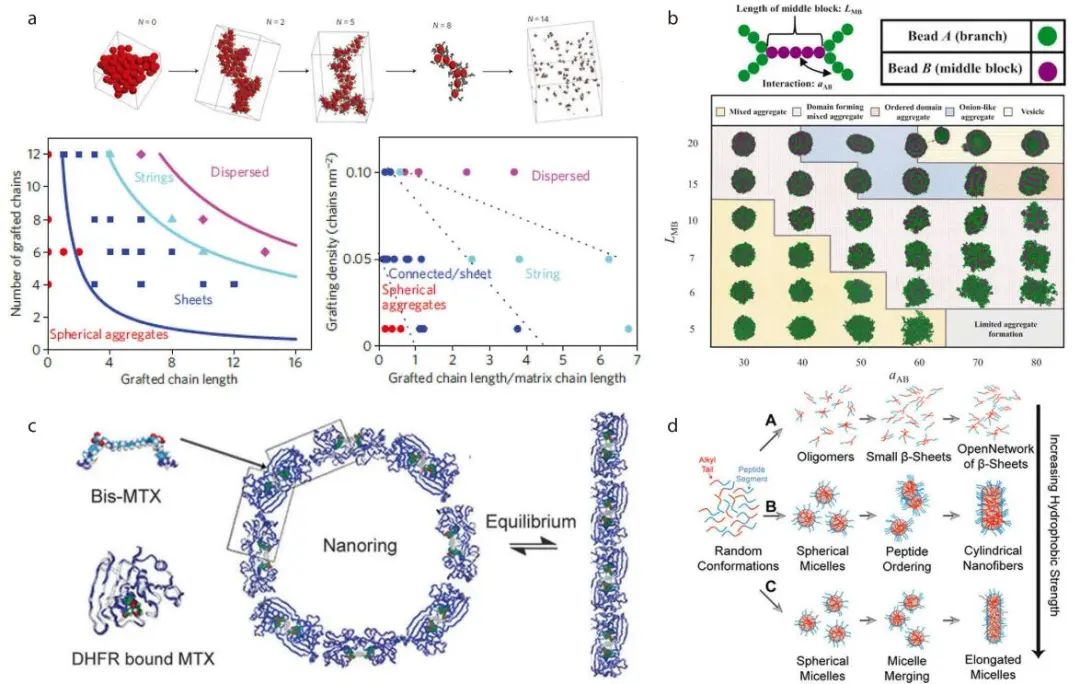
以接枝纳米粒子的自组装为例,将模拟结果与实验数据进行对比,证明了模拟结果的准确性和可靠性。
图4:模拟预测以及模拟与实验的比较
根据组成,接枝的纳米粒子可以微相分离成多种形貌。接枝链长度和接枝密度直接影响组装结构。通过MC模拟得到了接枝链长和接枝链数的相图,与实验结果一致
2、结晶
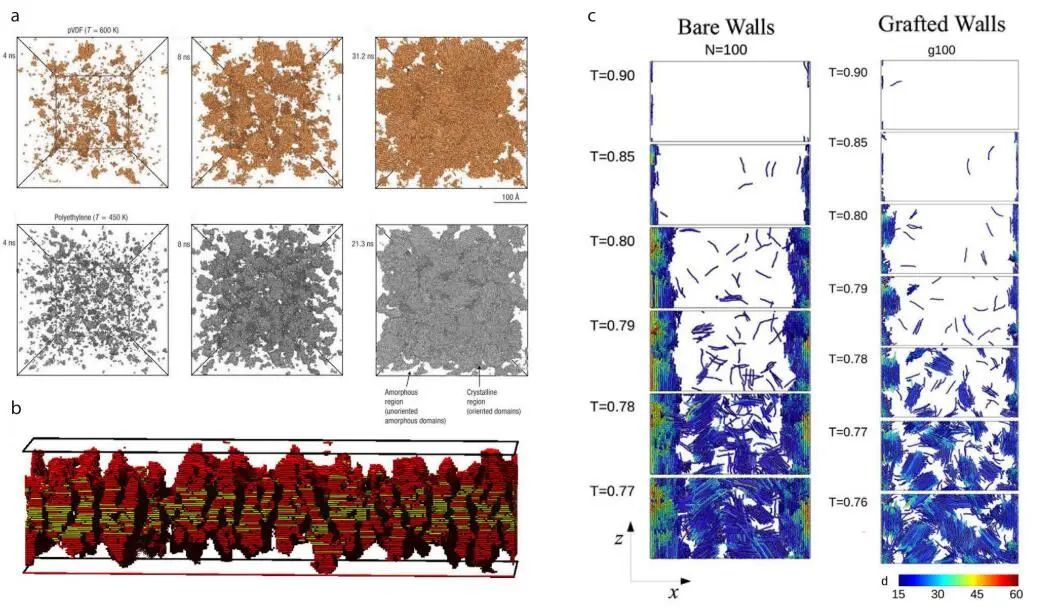
近一个世纪以来,研究者们一直在研究聚合物中错综复杂的结晶现象。模拟为聚合物结晶提供了一个独特视角,可以揭示结晶初期成核、生长和相关自由能垒的分子机制。
根据联合原子模型的MD模拟,当聚合物材料迅速冷却到相图的不稳定区域时,在聚合物材料中出现了调幅液-液相分离。
图5:结晶相关MD模拟快照
一般而言,MD和MC方法常用来研究结晶。MC方法可以提供以自由能表示的平衡状态,而MD方法可以显示结晶的动力学过程。
3、化学反应
化学反应是化学中最基本的课题之一,各种各样的模拟可以用来研究与之相关的问题。从头算被认为是通过从量子力学的角度在电子水平上描述化学反应来模拟电子转移等现象。
因此,探索包括中间状态在内的反应机理成为可能,这增强了对化学过程的理解。在MD模拟中创建了反应力场来表示化学反应,提供了键断裂和形成的精确描述。
图6:密度为1.227g/cm3的PDMS 3500K MD模拟的初始和最终配置
4、非平衡系统
复杂的非平衡问题也可以通过分子模拟进行研究。分子模拟可以用来观察和研究广泛的复杂非平衡过程。
然而,由于问题的复杂性以及严重依赖于现象的现实情况,最好是逐案确定合适的模拟方法。因此,进行非平衡模拟需要明确的研究目标和合适的模拟方法。
两大挑战 未来展望
本教程涵盖了分子模拟所必需的几个基本方面。随着计算机硬件和模拟技术的快速发展,分子模拟不仅可以解决基本的、常规的化学问题,还可以应用于各种复杂体系。
对于一个特定的科学问题,开始模拟的第一步是设计模拟体系,包括构建合理的初始模型和验证时空尺度。
下一阶段将重点研究模拟方法和最佳力场的选择。然后,综合考虑计算效率和方法,选择合适的软件。设置合适的参数后,最终准备运行模拟。
综上所述,对于分子模拟而言,力场的适用性、计算效率、模拟方法的准确性以及初始结构的合理性是决定模拟方案成功与否的主要因素。
目前,分子模拟的主要挑战之一是通过结合实验结果来提高模拟模型的精确构建与参数化。
分子模拟领域至今仍在经历着巨大的发展。另一个挑战是如何大幅度提高算法和软件的速度。
如果未来能在软件包的扩展和模拟技术的创建方面做出相当大的努力,那么研究人员进行分子模拟就必须更加方便。考虑到这一点,未来将有更多领域的关键问题通过分子模拟得到解决。

 咨询服务热线:
010-56245524
咨询服务热线:
010-56245524
 百强院校名师
百强院校名师  实战落地课程
实战落地课程  持续跟踪辅导
持续跟踪辅导  大量咨询案例
大量咨询案例